

Devic’s disease is a disorder of the central nervous system that strikes the nerves of the eye and spinal chord.
This disease occurs when the body’s immune system mistakenly attacks the cells of the nervous system, and although it mainly affects the optic nerve and spinal chord, it can also affect other parts of the body such as the brain.
In the past, neuromyelitis Optica was considered a type of multiple sclerosis, but with a different pattern of symptoms.
However, medical research has revealed the presence of certain antibodies in the body of patients with Devic’s disease, which are the key to obtaining a differential diagnosis of sclerosis.
Neuromyelitis Optica is a rare disease, more common in non-Caucasian populations, and is nine times more common in women than in men.
Between 80% and 90% of patients with this disease have flare-ups that leave sequels and can cause disability depending on the deterioration of the patient’s condition.
50% of patients lose sight in one or both eyes and may have the following symptoms:
- Weakness when walking;
- Painful cramps in the arms and legs;
- Paralysis of the legs. Patients will need assistance with walking during the first 5 years of illness;
- Hiccups and vomiting are uncontrollable;
- Loss of sensitivity;
- Disruption of the functioning of the bladder and intestines;
- Children with Devic disease may have seizures and sometimes coma.
Causes of Devic’s disease
Although the pathogenesis of Neuromyelitis Optica is not well understood, experts attribute this pathology to autoimmune diseases. NMO-IgG antibodies specific for this disease were identified in 2004. They are found in about 73% of patients and are not detected in patients with other autoimmune disorders, such as the typical form of multiple sclerosis, SEM, or other inflammatory lesions of the central nervous system. In 2005, the aquaporin-4 protein channel was discovered:
- It is a target for NMO-IgG antibodies.
- Aquaporin-4 is found mainly in the tissues of the spinal cord, in the brain – periventricular, and in the hypothalamus.
- Aquaporin-4 is localized in the processes of astrocytes and the walls of blood vessels that form the blood-brain barrier.
Damage to protein channels by NMO-IgG antibodies is accompanied by an increase in the permeability of the blood-brain barrier and free penetration of other immune complexes through it with the development of autoimmune inflammation.
- Morphologically, opticomyelitis is characterized by the development of necrosis of the white and gray medulla and the formation of demyelination zones;
- Pathological changes can be found in the spinal chord, optic nerves, hypothalamus, and chiasm;
- In the areas of demyelination and perivascular with opticomyelitis, IgG deposits are found;
- Chronic spinal inflammatory foci are accompanied by the development of cystic degeneration, atrophy, and gliosis;
- Sometimes the formation of cavities characteristic of syringomyelia is found;
- One of the components of morphological changes in opticomyelitis in many cases is autoimmune vasculitis.
Clinical manifestations of Devic’s disease
The main manifestations of OM are optic neuritis and myelitis. Lesions of the optic nerves and the spinal chord in some cases can occur simultaneously, but more often – with a time interval that can be months, years, or even decades.
Retrobulbar neuritis is often the first manifestation of the disease, preceding myelitis. The damage to the optic nerves is usually severe, can be unilateral or bilateral. With ophthalmoscopy, a normal picture of the fundus is found, or a slight blurring of the optic nerve discs and edema, in chronic cases – atrophy and pallor of the discs.
Myelitis in Devic’s disease has a severe course, with an acute development of symmetrical, gross motor, sensory and pelvic disorders. In 77-88% of patients after myelitis attack, partial restoration of motor functions occurs, however, complete regression is not guaranteed. In recurrent myelitis, the typical symptoms are para- or tetraparesis, paroxysmal muscle spasms, and radicular pain.
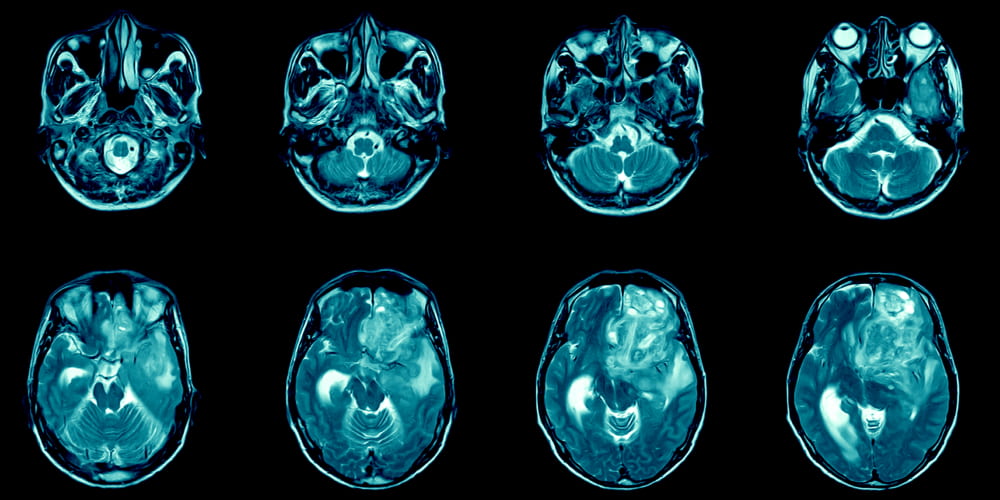
Among paraclinical diagnostic methods, MRI of the spinal cord is of the greatest importance in opticomyelitis. In most patients, MRI performed in the acute phase of myelitis reveals an extensive continuous lesion of the spinal chord that extends to more than 3 vertebral segments, it is swollen and edematous, and the lesion can accumulate a contrast agent, sometimes for several months. On MRI of the brain with Neuromyelitis Optica, either no pathological changes are detected, or nonspecific: asymptomatic and foci of demyelination are found.
The lesions in the brainstem and hypothalamus can also be considered characteristic and specific. Cerebral foci by their localization have a predisposition to those parts of the brain where there is a high level of immunoreactivity to AQP4 (aquaporin 4).
With an attack of myelitis in the analysis of cerebrospinal fluid, one-third of patients have pleocytosis (more than 50 leukocytes per 1 mm3) with the presence of neutrophils, an increase in protein levels.
In the blood serum of patients with OM, in almost half of the cases, various autoantibodies (antinuclear, extractable nuclear antigens, to double-stranded DNA, antithyroid) and their combinations are found, which indicates a predisposition of these patients to the development of autoimmune diseases.
A new step towards the diagnosis of OM was the discovery in 2004 in the plasma of patients with opticomyelitis of autoantibodies – NMO-IgG. which are specific biological markers of Devic’s disease. Since the sensitivity and specificity of this method are very high, the detection of autoantibodies in patients with symptoms of demyelinating disease allows a differential diagnosis between OM and multiple sclerosis, idiopathic transverse myelitis, recurrent or bilateral optic neuritis.
Devic’s disease diagnostics, diagnosis and treatment

Diagnostics
The clinical combination of optic neuritis and myelitis can be observed in typical multiple sclerosis, in systemic autoimmune diseases (systemic lupus erythematosus, Sjogren’s syndrome), in association with infectious processes (pulmonary tuberculosis, various viral diseases). It is impossible to reliably judge the presence or absence of OM based on clinical data alone.
In 2008, the Diagnostic Criteria for Neuromyelitis Optica (by: D. N. Miller) were adopted, which include:
- Large criteria (all basic criteria are required, but they can be separated by an indefinite time interval):
- optic neuritis with damage to one or both eyes;
- transverse myelitis with MRI-confirmed spinal cord lesion that extends to more than 3 vertebral segments on T2-weighted MRI images and is hypointense on T1-weighted images;
- lack of data for sarcoidosis, vasculitis, systemic lupus erythematosus, Sjogren’s syndrome, infectious process.
- Small criteria:
- a recently performed MRI of the brain should be free of pathology or detect nonspecific changes (foci in the dorsal regions of the medulla oblongata, foci in the hypothalamus and/or brain stem, “linear” foci located periventricular or in the corpus callosum, but not ovoid and not spreading into the parenchyma of the hemispheres of the brain);
- a positive test of blood serum or cerebrospinal fluid to NMO-lgG / antibodies to aquaporin 4.
Differential diagnosis
The differential diagnosis depends on the manifestation. With the classic picture, the diagnosis is quite reliable.
If the spinal cord is affected, it is necessary to carry out a differential diagnosis with lesions causing extensive transverse myelitis.
The defeat of the white matter of the cerebral hemispheres and the corpus callosum has a wide differential range, depending on the nature of the distribution of the lesions, but the most important in it is multiple sclerosis. The characteristic features suggesting opitcomyelitis and excluding MS are:
- periventricular distribution of foci in the area of the aqueduct;
- absence of perpendicularly oriented (along with the venules) periventricular foci (there are no foci like Dawson’s fingers);
- more extensive lesion of the corpus callosum;
- large, often confluent lesions;
- no contrast enhancement in the form of a half-ring;
- no damage to the gray matter of the cerebral cortex.
Treatment

There are conflicting data on the results of treatment with immunomodulatory and immunosuppressive drugs. To date, only 6 pharmaceuticals have been registered in the world that can be used in the pathogenetic therapy of opticomyelitis, 3 of which are interferons. However, their clinical efficacy has not been proven.
- To stop attacks of myelitis and neuritis, high doses of corticosteroids and plasmapheresis are used.
- With an intractable attack, it is possible to use rituximab or mitoxantrone.
- To prevent exacerbations, prednisone is used.
- Symptomatic therapy of Neuromyelitis Optica is based on the use of muscle relaxants, intrathecal infusion of baclofen, antidepressants, central analgesics, physiotherapeutic treatment.
- To restore the motor and sensory function of the limbs, improve coordination, reduce spasticity, the patient is shown physical therapy.
- For the treatment of an attack of opticomyelitis, high doses of corticosteroids (methylprednisolone 1000 mg per day, intravenously, for five consecutive days) are used.
- Supportive therapy with prednisolone 1 mg/kg per day is then recommended as part of initial immunosuppressive therapy to prevent recurrent attacks.
- Sometimes myelitis does not respond well to corticosteroid therapy; in such cases, plasmapheresis, immunosuppressive therapy, including oral prednisolone and azathioprine, is indicated. Over time, the dose of prednisolone is gradually reduced to the minimum maintenance dose or is canceled altogether, leaving only monotherapy with azathioprine.

 (150 votes, average: 4.82 out of 5)
(150 votes, average: 4.82 out of 5)





I've given up... the stress her office staff has put me through is just not worth it. You can do so much better, please clean house, either change out your office staff, or find a way for them to be more efficient please. You have to do something. This is not how you want to run your practice. It leaves a very bad impression on your business.
Please, leave your review
Write a comment: