

Huntington’s disease is an inherited genetic neurodegenerative brain disease with delayed manifestation. That is, the person inherits the mutant gene. But the manifestation of the disease itself can occur in each person at different ages. It means that the carrier or sick parent can have healthy or sick children.
A genetic mutation that affects the huntingtin protein causes HD. This protein plays an important role in the normal functioning of brain cells. The mutation causes the protein to become toxic and accumulate in the brain. This leads to cell death and ultimately to symptoms of HD.
There is currently no cure for Huntington’s disease. But there are treatments available to manage the symptoms and improve life quality. Doctors diagnose HD on the basis of:
- clinical examination;
- family history;
- genetic testing.
Advances in medical research have led to the development of new diagnostic tools. But, we need more research to find effective treatments. Read more about Huntington’s disease diagnosis and healing below.
What is Huntington’s Disease?
Huntington’s disease is a disease of the nervous system. The appearance of periodic muscle cramps or spasms is typical characteristic. The disease results from the gradual degeneration and death of nerve brain cells. The disease can develop at any age. But the earlier the first symptoms appear, the faster the disease progresses.
Different specialists treat Huntington’s chorea. All depends on:
- noted manifestations;
- prevail in the patient at a particular period of the disease’s progression.
Among the initial manifestations of the disease are sometimes noted:
- behavioral disorders;
- decreased sexual desire;
- gambling addiction;
- a tendency to excessively expression of emotions;
- the appearance of delirium;
- hallucinations.
This is the domain of psychiatrists.
In other cases, Huntington’s disease, without proper treatment appears, specifically with motor disorders – so-called hyperkinesias. These are involuntary movements in various body parts – the face and extremities. Neurologists are responsible for the treatment of these manifestations.
According to our observations, this disease is quite rare. In Caucasoids, the incidence ranges from 3 to 10 cases per 100,000 people. In other races, the incidence is lower – 1 per 1,000,000. At the same time, we must not discount the fact that Europeans may be simply examined more often. And thus, the diagnosis rate of Huntington’s disease is higher among them.
A person can get the disease through the autosomal dominant type. If one parent has the disease, there is a 50% chance of inheriting it. Both sexes can have the condition, but men are slightly more likely to have it. If a child gets the mutation from the father, the severity of the modification may increase. It means that children will develop the disease at an earlier age than the father.
Diagnosis of Huntington’s disease
Huntington’s disease is a genetic disease. That is, it is in the genes of the child. Thus, during intrauterine development, you can determine fetal disease. As a rule, the most revealing method of diagnosing Huntington’s disease is determining the number of CAG trinucleotide repeats in the Huntingtin gene:
- The number of repeats is less than 35. It means 100% confirmation that there is no possibility of developing the disease.
- If the number of repeats is greater than 36, it is an absolute fact of the inevitable development of the disease.
But no confirmation of the exact period of disease manifestation. The fact is that the age of onset of Huntington’s disease is quite individual:
- in some people, chorea begins to manifest in childhood;
- others, and this is the most common group, in adulthood;
- for others in old age.
But, because of our knowledge gained in practice, we see more Huntington’s disease therapies. Thus, the longer the polyglutamine tract, the greater the risk of early and severe manifestation. Below we can read more about the diagnosis of Huntington’s disease.
Prenatal genetic diagnosis
This is an invasive diagnosis of the fetus during intrauterine development. According to our practice, there are several methods of prenatal diagnosis:
- Chorion biopsy. It is usually performed in the early stages of pregnancy, from 10 to 14 weeks. Doctors take the cells from which the placenta is subsequently formed for analysis. The risk of complications in late pregnancy is 1% and does not exceed the population risk. At this stage, it is very important to take Huntington’s medication.
- Placenta-biopsy takes place between the 14th and 20th week of pregnancy. And doctors also take placenta cells for analysis. The risk of complications is 0.3%.
- Amniocentesis. Doctors perform it between the 15th and 18th weeks of pregnancy. They take the amniotic fluid, and for this analysis, they puncture the bladder. The risk of complications is 0.2%. This method really helps to cure Huntington’s disease.
- Cordocentesis takes place from the 20th week until the end of the pregnancy. It involves obtaining blood from the umbilical cord. The risk of complications for continued pregnancy is 3.3%.
- Fetal tissue biopsy. Doctors perform it from the 14th week. They take fetal tissue cells for analysis. But this is a rare diagnostic method.
Genetic method
Doctors use a genetic method for the diagnosis of Huntington’s disease in children or adults. It involves taking a blood sample from the patient. They determine the number of CAG repeats in each NTT allele. The number of repeats less than 35 is almost an absolute confirmation. This means that the patient will never develop Huntington’s disease. Moreover, you should understand that more than 36 times the number of repeats is not yet a diagnosis. The disease manifestation may be much later than the analysis performed. But it does mean that it (manifestation) will inevitably happen at some point. If there are similar symptoms, we are discussing a different disease.
Differential diagnosis
Differential diagnosis is crucial for definitive diagnosis. Almost always (over 90%), physicians must make a diagnosis based not only on:
- typical symptoms;
- family history of at least one blood relative with Huntington’s disease.
Doctors must have confirmation of genetic testing to cure Huntington’s disease. In all other cases, the condition will be different if the symptoms are similar. It will be Huntington’s cordiform disorder.
It is also worth saying that the presence of the mutant HD gene can also be in some other diseases. For example, one of the most common disorders is sporadic “senile” chorea. People often confuse it with Huntington’s disease when performing therapies. The following disorders are not accompanied by the increased number of trinucleotide repeats:
- benign familial chorea;
- abetalipoproteinemia;
- Alzheimer’s disease;
- hereditary ataxias and schizophrenia.
Thus, not only the presence of the mutant gene and the sign of heredity is very important.
Other instrumental diagnostic methods
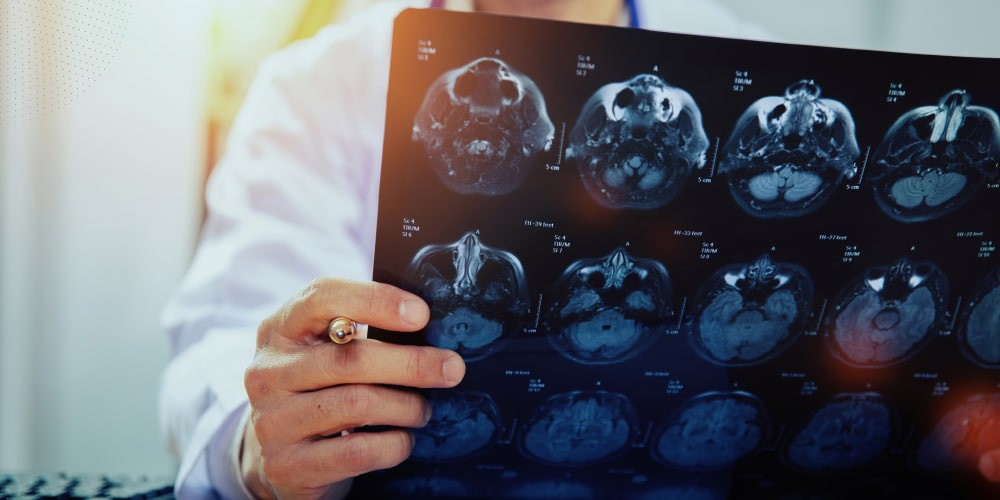
Positron emission tomography or functional magnetic resonance imaging detects changes in brain activity. These diagnostic methods can detect changes before the onset of the disease. But they are not specific to Huntington’s disease.
Computed tomography or magnetic resonance imaging is the best for further Huntington’s disease treatment. But they are relevant during the manifestation of the disease. They only show visible brain atrophy that is already present.
Huntington’s Disease Treatment
Treatment for Huntington’s disease is supportive and symptomatic. In fact, the focus is on eliminating motor symptoms and depression. As symptom correction improves the quality of life of the patient and his/her loved ones. The people closest to the patient, who are relatives and friends, give a great deal of help.
Research aimed at finding a specific treatment is ongoing. Still, researchers continue to look for ways to slow down and stop the progression of the disease. The European Association for the Study of Huntington’s Disease plays a significant role. Scientists are developing medication for Huntington’s disease on a mechanistic level. These include:
- strategies to reduce the expression of mutant proteins using antisense methods (in clinical trials);
- autophagy activation.
Antisense strategies use nucleic acid oligonucleotides. They have complementary sequences to the Huntington’s disease gene. Thus, they reduce the amount of gentingtin synthesized. This strategy is quite rational. Because the main driver of the disease is mutant gentingtin. There are other ways to cure Huntington’s disease. Read about them below.
Physical and occupational therapy
Physical therapy focuses on improving movement and function through:
- exercise;
- stretching;
- other methods.
The physical therapist can create an individualized exercise program. It will meet the needs and abilities of the patient. It focuses on developing balance, coordination, and muscle strength.
But, occupational therapy focuses on helping patients with daily living:
- dressing;
- bathing and cooking.
An occupational therapist can provide adaptive equipment and techniques. This will help patients perform these tasks more easily and safely.
Nutritional support
Nutritional support is important for Huntington’s disease treatment. Patients may have difficulty eating and maintaining weight due to some factors. These may include:
- motor symptoms that make swallowing difficult;
- cognitive and psychiatric symptoms that may affect appetite and food choices.
A balanced diet with adequate nutrition is essential for patients with HD. Because malnutrition can worsen symptoms and lead to other health problems. A nutritionist may recommend:
- dietary modification;
- supplements depending on the patient’s needs and preferences.
Huntington’s Medication
To relieve the condition, doctors use symptomatic treatment with Huntington’s medication. Some of the most effective ones we have seen are:
- Tetrabenazine is a drug specifically designed for symptomatic relief of Huntington’s disease.
- Antiparkinsonian drugs are appropriate to relieve muscle stiffness.
- Valproic acid is good for symptomatic relief of myoclonic hyperkinesia.
- Selective mirtazapine and serotonin reuptake inhibitors are excellent for depression.
- Atypical antipsychotics are appropriate for developing psychosis or impaired behavior or behavioral assessment.
Stages of Huntington’s disease
Huntington’s disease (HD) is a progressive neurodegenerative disease. It affects nerve cells in the brain. It leads to a gradual loss of motor, cognitive, and mental functions. The symptoms of HD usually progress throughout 10 to 25 years. According to our experience, we have grouped them into different stages. A human needs to have Huntington’s disease therapies starting with the early stage. They are usually based on the severity of the symptoms.
The stages of HD are as follows:
- Early Stage. This stage is characterized by subtle changes in movement, such as clumsiness, difficulty with balance, and involuntary jerking movements (chorea). Cognitive symptoms may also appear in this stage. It can be difficult with planning, organizing, and problem-solving.
- Middle Stage. In this stage, motor symptoms become more pronounced. And patients may experience difficulty with walking, speaking, and swallowing. Cognitive and psychiatric symptoms also become more severe, with patients experiencing memory loss, confusion, depression, and anxiety.
- Late Stage. This stage is usually marked by severe motor and cognitive impairment, including a loss of speaking, walking, and swallowing ability. Patients require full-time care and may experience hallucinations, delusions, and other psychiatric symptoms.
The progression of HD can vary between individuals. It can be affected by several factors, including age of onset, genetic factors, and overall health. Huntington’s disease therapies focus on managing symptoms and improving the quality of life for patients at each stage of the disease.
If you or a loved one is living with Huntington’s disease, Lone Star Neurology is here to help with Huntington’s medication. Our team of experienced neurologists specializes in diagnosing and treating neurodegenerative disorders. We offer a comprehensive range of services. It includes:
- genetic counseling;
- clinical evaluation;
- personalized treatment plans tailored to each patient’s unique needs.
Our team of caring professionals is dedicated to providing compassionate, patient-centered care. Our state-of-the-art facilities have the latest technology to provide an accurate and timely diagnosis of Huntington’s disease. Contact us today to learn more about our services. So we can help you or your loved one with Huntington’s disease.
FAQ
- Can Huntington’s disease be treated?
You can manage its symptoms with medications and therapies. Supportive care is also good for improving the quality of life for patients. But there is currently no cure for Huntington’s disease.
- What triggers Huntington’s disease?
A genetic mutation in the huntingtin gene causes Huntington’s Disease. Because the body produces a toxic protein that damages brain cells. Humans inherit this mutation in the autosomal dominant type.
- What are the 5 stages of Huntington’s disease?
The 5 stages of Huntington’s disease are premanifest, early stage, middle stage, late stage, and end-of-life stage. These stages are based on the severity and progression of the disease.
- How does Huntington’s disease manifest itself?
Dementia and psychiatric disorders are the main symptoms. The patient may experience depression, apathy, irritability, anhedonia, antisocial behavior, and bipolar or schizophrenic-like disorders.
 (No Ratings Yet)
(No Ratings Yet)





I've given up... the stress her office staff has put me through is just not worth it. You can do so much better, please clean house, either change out your office staff, or find a way for them to be more efficient please. You have to do something. This is not how you want to run your practice. It leaves a very bad impression on your business.
Please, leave your review
Write a comment: